[질의응답]인보사케이주 조사 관련 식약처 Q&A
-
기사 스크랩
-
공유
-
댓글
-
클린뷰
-
프린트
"신장 세포로 바뀐 경위 및 사실관계를 정확히 확인하기 위해 시험검사(2019.4.9∼5.26), 추가자료 제출(2019.4.15∼5.14) 및 검토(2019.5.15∼), 현장조사(2019.5.2, 5.8, 5.10), 美 현지실사(2019.5.20∼24) 등의 검증과정이 필요했기 때문임. 특히 최초 세포의 경우 미국에서 한국으로 운송(2019.4.16.한국도착), 세포배양(2019.4.17∼5.2)에 시간이 소요됐고, 세포사멸시험의 경우에는 한 달 반의 시간(2019.4.11∼5.26)이 걸렸음."
▶허가 당시 중앙약사심의위원회를 2회 개최한 사유는?
"인보사케이주의 허가과정 중에 실시한 1차 중앙약심 자문결과(2017.4)와 3상 임상시험 계획 승인 시 실시한 중앙약심 자문결과(2013.7)가 서로 상충됐음. 3상 임상시험 계획 승인 시 실시한 중앙약심에서는 골관절염치료제로서 연골구조 개선이 없더라도 관절기능 및 통증 개선(1년 이상 관찰)을 보인다면 유전자치료제의 유효성으로 적절하다고 보아, 임상시험 계획을 승인(2013.8)했음. 그러나 1차 중앙약심에서는 골관절염의 유전자치료제는 연골의 구조개선 없이 증상완화(기능 및 통증 개선)만으로는 유효성을 인정할 수 없다고 자문함. 이에 따라 1차 중앙약심의 참석위원과 3상 임상시험 계획을 승인한 위원을 모두 포괄해 제품의 유효성에 대한 종합적인 자문을 받고자 2차 중앙약심을 개최함(2017.6)."
▶2차 중앙약심위원 구성시 일부 위원을 배제했다는 것이 사실인지?
"2차 중앙약심위원은 제품의 유효성에 대한 이견 해소 및 종합적인 자문을 받기 위해 포괄적으로 구성함. 이를 위해 1차 중앙약심 위원 전원과 3상 임상시험 계획 승인을 위한 중앙약심 참석 위원 일부를 위촉했음. 위원회 정족수를 채우기 위한 신규 위촉이 있었을 뿐, 이 과정에서 특정위원 배제는 없었음."
▶세포가 바뀐 사항을 GMP 등 사후관리에서 발견하지 못한 이유는?
"의약품 사후관리는 허가단계에서 확립한 품질기준에 따라 일관성을 갖고 제조하고 있는지를 점검하는 것. 연구개발 단계에서 발생한 문제를 제조품질관리(GMP) 등 사후관리를 통해서는 발견하기는 현실적으로 어려움. 이러한 문제를 보완하기 위해, 허가시 제출자료의 신뢰성 검증을 강화하는 등 제도를 개선하겠음."
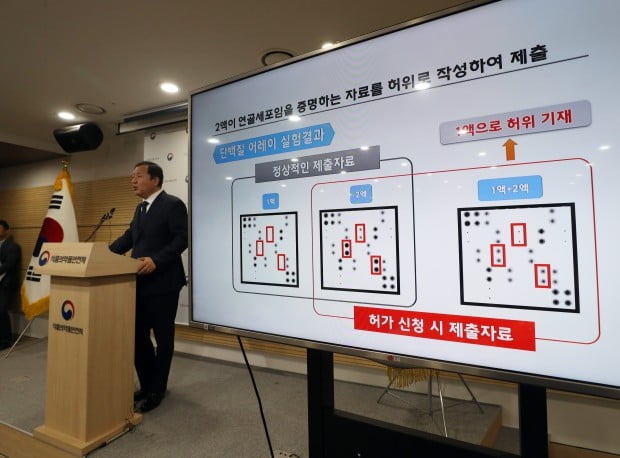
▶2액이 신장세포로 확인됐는데 안전한 것인지?
"식약처의 세포사멸시험을 통해 44일 후 세포가 더 이상 생존하지 않음이 확인됐음. 또 국내에서 실시한 임상시험 결과, 임상시험 참여자에 대한 장기추적관찰 및 시판 후 수집된 안전성 자료에서도 약물과 관련된 중대한 부작용 사례는 없었음. 이러한 결과 및 독성 및 임상자료에 대한 전문가 자문(2019.4.9∼ 4.11) 등을 종합적으로 검토해 볼 때 현재까지는 안전성 측면에서 큰 우려가 없는 것으로 판단됨. 다만 2액이 연골세포가 아니라 신장세포로 확인됨에 따라 만약의 경우 발생할 수 있는 부작용에 대비하기 위해 전체 투여환자(438개 병의원 3707건 투여)에 대한 특별관리와 15년간 장기 추적조사를 실시할 예정."
▶장기추적조사 진행 상황은?
"현재 총 투여환자 중 1040명이 약물역학 웹기반 시스템에 등록됨. 오는 10월까지 등록을 완료하고 15년 간 장기추적조사 시행 예정."
▶현재까지 발생한 부작용 현황은?
"현재까지 수집된 이상사례 분석 결과, 약물과 연관된 중대한 부작용 사례는 없음. 시판 후 보고된 주요 이상사례 311건은 주사부위반응(62건), 주사부위통증(61건) 등 주로 국소적으로 나타는 부작용임. 이는 의약품과의 인과관계와 관계없이 보고된 것으로서, 이 자료만으로는 특정 제품에 의해 부작용이 발생하였다고 확정할 수 없음. 종양 관련한 이상사례로는 위암종 등 4건이 보고됐으나, 약물과의 인과관계가 없는 것으로 확인됨."
한민수 한경닷컴 기자 hms@hankyung.com
관련 뉴스
-
1
식품의약품안전처는 28일 코오롱생명과학의 퇴행성관절염 유전자치료제 '인보사케이주'의 판매허가를 취소한다고 밝혔다. 주요 성분이 허가 당시 제출한 자료에 기재된 연골세포가 아닌 신장세포로 확인됐기 때문이...
![[속보] 식약처, 코오롱생명과학 '인보사' 허가 취소](https://img.hankyung.com/photo/201905/02.19752778.3.jpg)
-
2
허위자료 제출·사실은폐 정황 드러나…성분 바뀐 경위·근거 제시 못 해 코오롱생명과학·코오롱티슈진, 2017년 의약품 성분 뒤바뀐 사실 인지 정황 식약처 "현재까지 환...
-
3
식품의약품안전처가 28일 오전 의약품 성분이 바뀐 골관절염 유전자치료제 '인보사케이주(인보사)'에 대한 조사 결과와 행정처분 수위를 발표한다.인보사는 사람 연골세포가 담긴 1액과 연골세포 성장인자를 도...