95년부터 "의약품 재심사제도" 시행...보사부
위해 오는 95년 1월1일부터 "의약품 재심사제도"를 도입, 실시하기로 했다.
새로 실시될 이 제도는 신약으로 제조허가를 받은 모든 의약품에 대해
6년이 경과한 날로 부터 3개월이내에 그동안의 시판과정에서 나타난
부작용,임상결과 등을 토대로 유효성 및 안전성에 관한 자료를 의무적으로
작성,제출토록 해재심사를 실시하는 것을 골자로 하고 있다.
보사부는 재심사 결과 약효가 떨어지고 안전성에 문제가 있는 것으로
판명될 경우 품목제조허가나 품목변경등의 행정조치를 취하고 재심사 대상
품목에는 수입의약품도 포함시키기로 했다.
보사부는 신약을 재심사하는 동안 다른 업체에서 동일 품목을
제조하고자할 경우에는 최초 제조허가자가 신약허가시 제출했던 동일한
임상자료를 제출토록 했다.
신약이란 대한약전이나 공정서에 올라있지 않은 것으로 국내에서 이미
허가된 의약품과는 화학구조 또는 본질조성이 다른 신물질과 이를
유효성분으로 함유한 의약품을 말한다.
신약제조 허가시 필요한 첨부자료로는 <> 기원,발견 및 개발경위에
관한 자료 <>구조,결정,물리화학적 성질에 관한 시험자료 <> 안전성에
관한 자료 <> 독성에 관한 시험자료 <> 효력시험에 관한 자료 <>
일반약리에 관한 자료<>흡수,분포,대사 및 배설에 관한 시험자료 <>
임상시험 성적자료(3개이상 임상시험 지정의료기관에서 각 30예 이상)등이
있다.
-
기사 스크랩
-
공유
-
프린트
-
1
19일 서울 견지동 한국불교역사문화기념관에서 열린 ‘화암사 사리 이운 고불식’에서 스님들이 부처님과 고려시대 고승들의 사리를 친견하고 있다. 대한불교조계종은 지난 16일 미국 보스턴박물관을 방문해 가섭불·석가모니·정광불 및 고려시대 스님인 나옹선사(1320∼1376)·지공선사(?∼1363)의 사리 등을 돌려받았다. 연합뉴스
![[포토] 美 보스턴서 돌아온 사리 친견하는 스님들](https://img.hankyung.com/photo/202404/AA.36471428.3.jpg)
-
2
롯데웰푸드, 태국 면세점 입점…빼빼로 앞세워 관광객 공략
롯데웰푸드는 대표 브랜드인 ‘빼빼로’와 ‘제로’가 태국 면세점 킹파워에 입점했다고 19일 밝혔다. 국내 제과업체 제품이 태국 면세점에 입점한 것은 이번이 처음이다.롯데웰푸드는 지난 13~15일 열린 태국 전통문화 축제 ‘송끄란’에 맞춰 빼빼로 세트 2종과 제로 2종(후르츠 젤리, 크런치 초코볼)을 킹파워 면세점에서 판매해 홍보 효과를 높였다. 킹파워는 태국에서 가장 규모가 큰 면세점이다. 1989년 설립돼 현재 방콕 돈므앙국제공항, 월드트레이드센터 등에서 면세 사업을 하고 있다.태국은 연간 4000만 명 이상의 해외 관광객이 방문하는 나라다.롯데웰푸드 관계자는 “이번 입점은 빼빼로와 제로를 글로벌 관광객에게 알리는 계기가 될 것”이라고 말했다.하헌형 기자
-
3
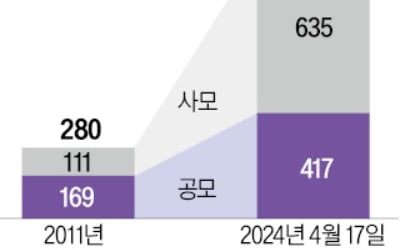
미국 호주 등 자본시장 선진국의 공통점은 공모펀드 시장이 튼튼하다는 것이다. 전문가에게 돈을 맡겨 돈을 불리는 펀드가 ‘증시 안전판’ 역할을 하면서 가계 자산 증식과 기업 성장으로 이어지는 선순환 구조가 잘 구축돼 있다. 한국 펀드시장도 지난 몇 년간 고성장했지만, 과실은 국민들에게 고루 돌아가지 못했다. 일부 고액 자산가만 가입할 수 있는 사모펀드 시장으로 성장축이 기울어진 탓이다.19일 금융투자협회에 따르면 지난 17일 기준 국내 전체 펀드 순자산액은 1052조원으로 집계됐다. 2017년 순자산총액 500조원을 달성하기까지 47년이 걸렸지만, 7년 만에 두 배인 1000조원을 넘어섰다.펀드시장의 급성장은 사모펀드가 이끌었다. 사모펀드 규모는 10년 전 176조원에서 현재 635조원으로 260% 늘었다. 같은 기간 공모펀드 순자산은 198조원에서 417조원으로 110% 증가하는 데 그쳤다. 공모와 사모의 비중은 2014년까지만 해도 5.3 대 4.7로 공모가 높았지만, 현재는 3.7 대 6.3으로 역전됐다.사모펀드는 공모펀드보다 상대적으로 규제가 적기 때문에 메자닌, 선물옵션, 부동산 대체 등 다양한 전략을 사용한다. 하지만 서민에겐 ‘그림의 떡’이다. 전문투자자가 아닌 일반투자자가 사모펀드에 가입하려면 최소 3억원의 현금이 필요하고 판매사에 따라선 전문투자자가 아니면 아예 받지 않는다. 전문투자자가 되려면 최근 5년 중 1년 이상 월말 평균잔액 5000만원 이상, 금융투자상품 계좌 개설 1년 이상 등 까다로운 조건을 갖춰야 한다.정작 국내 증시의 안전판 역할을 해야 할 국내 주식형 액티브 공모펀드는 설정액 규모가 5년 전 24조308억원에서 현재 13조8233억원으로 매년 쪼그라들고 있다. 국내 주식

![넷플릭스, 가입자 순증 꺾였다…악재 쏟아진 기술주 [글로벌마켓 A/S]](https://timg.hankyung.com/t/560x0/photo/202404/B20240419072033320.jpg)